Neuropatías genéticamente determinadas
ENFERMEDAD DE CHARCOT-MARIE-TOOTH
1. Introducción:
Enfermedad neuromuscular hereditaria más frecuente. Se han identificado más de 25 genes implicados, todos ellos con un fenotipo clínico final común de degeneración axonal.
2. Clínica:
Debilidad muscular de inicio en miembros y de predominio distal junto con pérdida de sensibilidad de progresión disto-proximal. La pérdida de sensibilidad es principalmente táctil, vibratoria y nociceptiva. La perdida de sensibilidad propioceptiva puede dar lugar a ataxia.
El debut es en las primeras décadas de la vida con progresión lenta.
Los síntomas se inician en los pies, con aumento del arco plantar, dedos en martillo y debilidad de musculatura intrínseca, ascendiendo progresivamente a la pierna y tercio inferior de muslo. Es en esta fase cuando se inicia la afectación de la mano y el antebrazo.
Los reflejos osteotendinosos disminuyen o desaparecen de manera ascendente.
Otros síntomas: deformidades ósea como pie cavo, dedos en martillo o escoliosis; dolor, calambres musculares, temblor en manos, acrocianosis…( Cruse R; 2007)
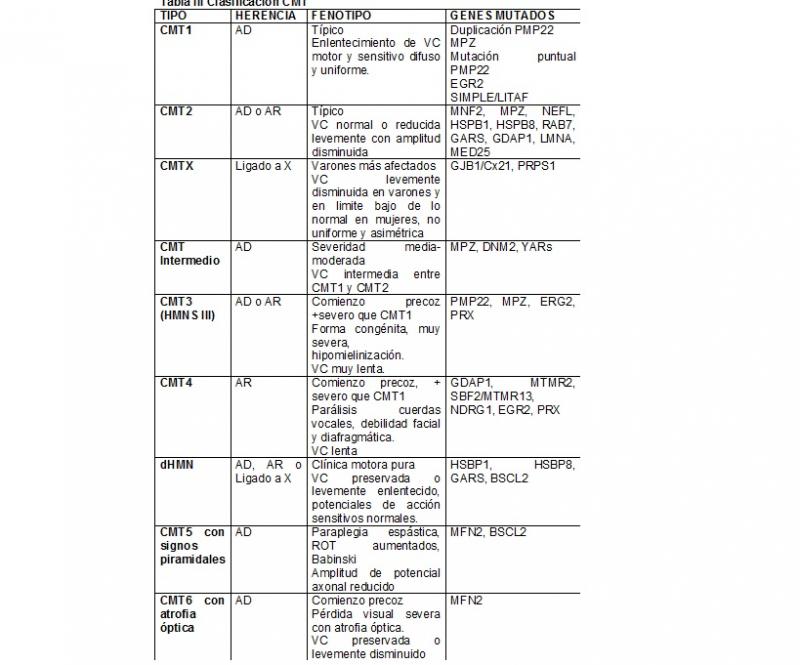
3. Clasificación [Tabla I ] (Pareyson D;2009)
- Desmielinizante:
CMT1 y CMT4: caracterizada por velocidad de conducción disminuida en el ENG y anomalías en la mielina en la biopsia de nervio.
Subtipo desmielinizante más severo: CMT3 o Neuropatía Déjèrine- Sottas
- Axonal:
CMT2: velocidad de conducción normal o levemente disminuida, disminución de amplitud de los potenciales, evidencia en la biopsia de degeneración y regeneración axonal.
- Formas intermedias entre las dos anteriores:
Dominantes: DiCMT y ligadas a X: CMTX1.
- Formas motoras puras:
dHMN (neuropatía hereditaria motora distal)
4.Genética
- Autosómico dominante: la más frecuente en CMT 1 y la mayoría en CMT2
- Autosómico recesivo: en CMT 4, CMT 2 y dHMN
- Ligadas al X dominante: en CMTX1
- Ligadas al X recesivo: en CMTX2, CMTX3, CMTX4 y CMTX5
- De novo: en CMT1A y CMT2A (Colomer; 2008)
5. Diagnóstico
5.1.- Estudios neurofisiológicos: en función de la velocidad de conducción:
- Muy disminuida: CMT1, CMT3 y CMT4
- Normal o levemente disminuida con reducción de la amplitud de los potenciales de acción sensitivos y motores: CMT2
- Disminuida de manera intermedia con potenciales de acción normales: CMTX1 en varones o Di CMT
- Normal o casi normal con potenciales sensitivos normales: dHMN
5.2.- Tests moleculares: se orientarán los test genéticos en función del subtipo que se sospeche y la frecuencia de aparición de mutaciones de cada gen en cada subtipo.
5.3.- Biopsia de nervio: limitada a casos esporádicos o casos familiares en los que los test moleculares han dado negativos. ( Cruse R; 2007)
6. Tratamiento
No existe tratamiento específico, solo sintomático
NEUROPATÍA RECURRENTE POR SUSCEPTIBILIDAD A LA PRESIÓN
Neuropatía Tomacular o neuropatía familiar con susceptibilidad a la parálisis por presión
1. Definición
Neuropatía sensitivomotora recurrente en un nervio aislado de herencia AD.
2. Prevalencia
2-5 casos /100.000
3. Clínica
El síntoma más común es una mononeuropatía aguda no dolorosa, con antecedente o no de compresión del nervio, de inicio en la adolescencia o adulto joven. La parálisis del nervio puede recurrir en los siguientes años pero algunos individuos solo tienen un episodio o permanecen asintomáticos. El 50% logran la recuperación completa y los síntomas persistentes no suelen ser muy severos.
Los nervios más afectados son: el N. peroneo (pie caído), N. cubital (debilidad interósea e hipotenar), N. mediano (S. del túnel carpiano), plexo braquial y N. radial.
4. Diagnóstico
De sospecha en un paciente con neuropatía compresiva recurrente con historia familiar positiva para herencia AD. Para confirmar el diagnóstico se requiere la detección de la mutación en el gen PMP22.
5. Tratamiento
Preventivo y de las manifestaciones clínicas. (Bird TD; 2010)
NEUROPATÍAS PORFÍRICAS
Las porfirias aguda intermitente, variegata y coproporfiria hereditaria pueden cursar con neuropatía. Todas ellas tienen una herencia AD.
1. Clínica.
La neuropatía periférica puede aparecer de forma aguda en las exacerbaciones de la porfiria o de forma progresiva. Se afectan primero las extremidades superiores, con debilidad y parestesias o temblor de acción. Los síntomas sensitivos son de predominio proximal, en “bañador”, afectando nalgas y hombros. Pueden existir alteraciones autonómicas y alteraciones del comportamiento.
2. Diagnóstico:
En los ataques agudos, aparecen los precursores (porfobilinógeno, delta-aminolevulínico) y elevación de catecolaminas en orina; la coproporfirina está elevada en heces. LCR: hiperproteinorraquia. VC motora y sensitiva: suelen ser normales. Biopsia de nervio: degeneración axonal y desmielinización segmentaria.
3. Tratamiento.
No existe terapia específica.
ENFERMEDAD DE REFSUM
1. Definición
Enfermedad peroxisomal que cursa con polineuropatía sensitivomotora, debido al depósito de ácido fitánico en los tejidos.
2. Etiología
Causada por la deficiencia de la enzima fitanoíl-CoA hidroxilasa que cataliza el primer paso de la αoxidación del ácido fitánico.
3. Genética
Causada por mutación en el gen PHYH y en menor número en PEX7.
4. Clínica
Se caracteriza por la presencia de: retinitis pigmentaria, polineuropatía periférica, ataxia cerebelosa, e hiperproteinorraquia sin aumento celular. Debut entre la primera infancia y la 2ª década. Puede asociar ictiosis, sordera, arritmias y cardiomiopatía.
La polineuropatía es simétrica, crónica y progresiva, con pérdida de ROT, afectación del sentido postural y vibratorio, parestesias y dolores neuropáticos espontáneos.
5. Diagnóstico
- LCR. Hiperproteinorraquia (menor en las fases de remisión y mayor en las exacerbaciones).
- EMG. Signos de denervación. VC: muy disminuida en extremidades. Varía proporcionalmente a la remisión clínica y a la disminución del fitánico en sangre.
- Biopsia de nervio. Disminución de fibras mielínicas y formaciones en “bulbo de cebolla”. Al microscopio electrónico se detectan depósitos de ácido fitánico.
- Diagnóstico de confirmación. Aumento del ácido fitánico en sangre, orina y LCR.
6. Tratamiento
Dieta con restricción de ácido fitánico: aumenta la supervivencia y disminuye el riesgo de ceguera y de miocardiopatía. El riesgo neurológico se correlaciona con los niveles de ácido fitánico.
BIBLIOGRAFIA:
- Bird TD Hereditary Neuropathy with Liability to Pressure Palsies. (2010). Obtenido el 10 de abril de 2012 en www.uptodate.com.
- Colomer J. Polineuropatías sensitivo-motoras. . Madrid. Asociación española de Pediatria 2ªed.(2008). Obtenida el 10 de abril del 2012 en http://www.aeped.es/protocolos-neurologia.
- Cruse,R. Overview of hereditary neuropathies. (2007) Septiembre, 8.Obtenido el 10 de abril de 2012 en www.uptodate.com.
- Cruse,R. Hereditary primary motor sensory neuropathies, including Charcot-Marie-Tooth disease.2010 Septiembre, 8.Obtenido el 10 de abril de 2012 en www.uptodate.com
- Pareyson D, Marchesi C. (2009) Diagnosis, natural history, and management of Charcot-Marie- Tooth disease. Lancet Neurol; 8: 654-67.

{kind=link}