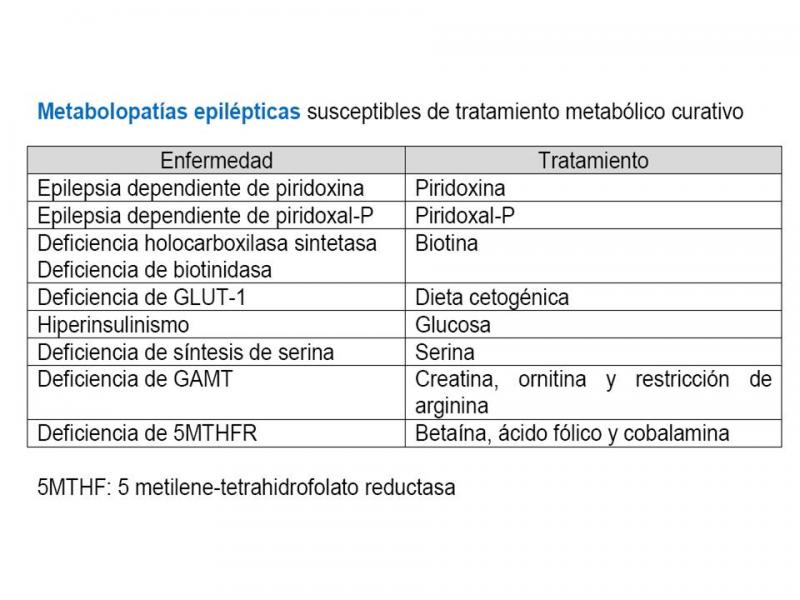
Metabolopatía neonatal con convulsiones
INTRODUCCION
Numerosos errores congénitos del metabolismo (ECM) pueden presentar crisis epilépticas en los primeros años de vida (aunque rara vez las crisis son la manifestación más importante). De todas maneras es preciso recordar que las causas que provocan crisis en los recién nacidos no suelen ser de origen metabólico: hipoxia, hemorragia cerebral, infección, patología cardiopulmonar, malformaciones cerebrales (Van Hove et al; 2011). El reconocer a una ECM como causa de epilepsia es fundamental no sólo para un correcto consejo pronóstico y genético, sino porque en algunos casos es posible un tratamiento específico curativo (Parish et al;2009).
Por tanto, ante un recién nacido con crisis epilépticas de causa desconocida hay que valorar iniciar tratamiento con:
- Piridoxina 100-500 mg iv y posteriormente 30 mg/kg/dia cada 8 horas durante 3 dias oral
- Piridoxal fosfato 30 mg/kg oral cada 8 horas durante 3 días.
- Ácido folínico no indicado.
- Biotina: 5-20 mg/dia.
CLAVES DIAGNÓSTICAS
La mayoría de las veces la presentación es inespecífica, aunque en ocasiones el patrón electroencefalográfico o el tipo de crisis pueden orientarnos en el diagnóstico:
1.- Patrón electroencefalográfico de brote-supresión (“burst-suppression”): se caracteriza por brotes de ondas lentas e irregulares con puntas imbricadas (burst) que se alternan de manera pseudo-periódica con fases de aplanamiento de la actividad (suppression) (Martínez-Bermejo et al;2001).
Se puede presentar en las siguientes entidades:
- Epilepsias dependientes de piridoxina y piridoxal-fosfato.
- Déficit del cofactor de molibdeno.
- Trastorno del ciclo de la urea: citrulinemia.
- Aminoacidopatías: leucinosis, hiperglicinemia no cetósica.
- Trastornos de la biotina: déficit de holocarboxilasa.
- Acidurias orgánicas: propiónica, metilmalónica, isovalérica, D2OHGA y D-glicérica.
- Peroxisomales: síndrome de Zellweger.
- Trastorno de las purinas: déficit de adenilosuccinato liasa.
- Otros: defecto de transportador mitocondrial del glutamato, síndrome de Smith-Lemli-Opitz.
2.- Los diversos tipos de crisis que aparecen son raramente específicos de un determinado tipo de ECM con la excepción de las crisis mioclónicas, especialmente si se presentan en forma de epilepsia mioclónica progresiva. Se puede presentar en las siguientes entidades:
- Epilepsia dependiente de piridoxina y piridoxal-fosfato
- Hiperglicinemia no cetósica.
- Defecto del cofactor de molibdeno.
- Déficit de biotina.
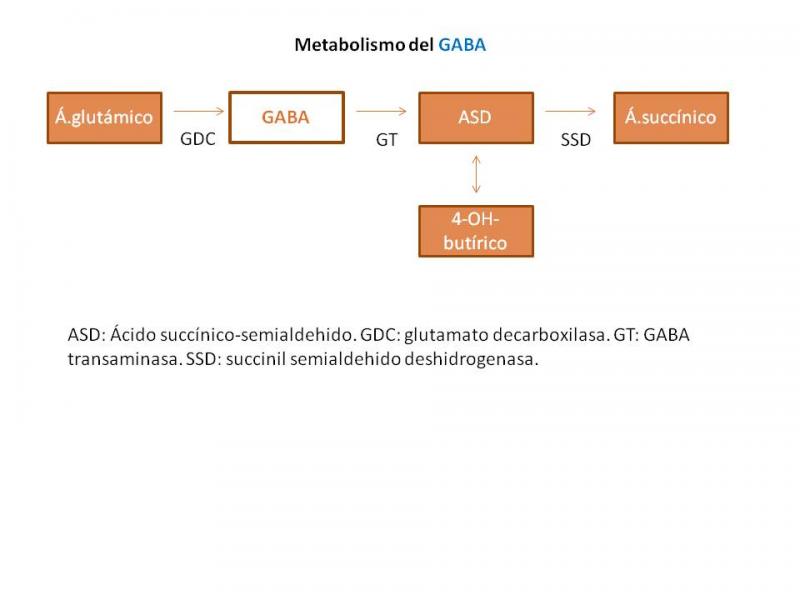
- Enfermedades de los neurotransmisores: déficit de GABA transaminasa.
- Enfermedades mitocondriales.
- CDG.
- Déficit en el metabolismo de las purinas.
- Enfermedades lisosomales.
ECM EN LOS QUE LA EPILEPSIA ES EL SINTOMA PRINCIPAL
- Epilepsia dependiente de piridoxina
Trastorno autosómico recesivo con gran heterogeneidad clínica causado por deficiencia de antiquitina (alfa-aminoadípico semialdehído deshidrogenasa), en la vía cerebral de degradación de la lisina. Esta alteración produce aumento de alfa-aminoadípico semialdehído (AASA) que se encuentra en equilibrio con el ácido pipereidin-6-carboxílico (P6C). El P6C se condensa con el piridoxal fosfato y lo inactiva. También aumenta el ácido pipecólico.
- Clínica
En las formas precoces de presentación neonatal, típicamente las crisis comienzan en los primeros días de vida incluso intraútero. Suelen ser frecuentes, breves y de todo tipo (especialmente mioclónicas o tónicas), siendo común el status convulsivo. Las crisis no suelen ocurrir de manera aislada sino que el paciente presenta un estado encefalopático (irritabilidad, sobresaltos) con manifestaciones sistémicas (alteración de temperatura, distensión abdominal, vómitos, distrés respiratorio y acidosis metabólica) (Baxter et al; 2001).
- Diagnóstico
Diagnóstico de sospecha: hace unos años se basaba en la demostración de la corrección de las crisis y mejoría del patrón EEG tras la administración de piridoxina; y la recurrencia de las mismas al retirar la piridoxina. Actualmente existen marcadores de enfermedad: elevación de ácido pipecólico y alfa-amino-adípico semialdehido deshidrogenasa (AASA) en sangre, orina y LCR.
Diagnóstico de confirmación: mutaciones en el gen de la antiquitina ALDH7A1.
- Pruebas complementarias:
EEG: en muchos casos brote-supresión o patrón multifocal. A veces puede ser normal, sobre todo en las formas atípicas.
Neuroimagen: pueden objetivarse anomalías estructurales cerebrales como leucoencefalopatías o hipoplasia del cuerpo calloso y del cerebelo. En pacientes con crisis no tratadas puede desarrollarse hemorragia intraventricular y/o subaracnoidea.
- Tratamiento
Según Plecko se recomienda administrar piridoxina 30 mg/k/d iv durante 3 días consecutivos. Otra opción es administrar 15 mg/kg/d por vía oral durante al menos 1 semana. En neonatos, Gospe recomienda una dosis inicial de 100 mg y, si no hay mejoría en 10 minutos, dosis adicionales de 100 mg hasta completar 500 mg (Gospe et al; 1998).
Si se obtiene respuesta se debe iniciar tratamiento de mantenimiento a 15-18 mg/kg/día (máximo 200 mg/día en niños pequeños y 500 mg/día en adultos). Se debe vigilar en el momento agudo la aparición de marcada depresión del sistema nervioso central, incluyendo apneas (Plecko et al; 2007).
También se han descrito pacientes con crisis epilépticas que responden al tratamiento con piridoxina (piridoxín-respondedores) sin presentar el trastorno metabólico. El pronóstico cognitivo depende de la edad de presentación, del retraso en el tratamiento, del tipo de mutación genética y de las malformaciones asociadas.
- Epilepsia dependiente de piridoxal-fosfato.
Alteración en la producción de piridoxal fosfato (cofactor en la síntesis de numerosos neurotransmisores).
- Clínica
Las primeras descripciones se realizaron en recién nacidos, generalmente con antecedentes perinatales (prematuros con APGAR bajo, acidosis metabólica o que requirieron ventilación mecánica), que presentaron epilepsia refractaria desde los primeros días de vida con gran componente mioclónico; que no respondían a piridoxina pero que sí lo hacían con la administración de piridoxal-fosfato (Hoffman et al;2007). Posteriormente, Hoffmann amplía el espectro clínico de la deficiencia de piridoxal fosfato al comunicar 6 nuevos casos con hallazgos, al menos en parte, no característicos, tanto en los datos clínicos, EEG y de laboratorio.
- Pruebas complementarias
EEG: brote-supresión.
- Diagnóstico
Diagnóstico de sospecha: respuesta a piridoxal fosfato. Indirectamente se puede objetivar en LCR una disminución de 3-metoxitirosina, ácido homovalínico (HVA) y 5-OH-indolacético (5-HIAA), así como un aumento de treonina y glicina. En orina es posible detectar aumento de vanil-láctico. Diagnóstico de confirmación: detección de mutaciones en el gen PNPO.
- Tratamiento
Se recomienda tratamiento de prueba con piridoxal-fosfato vo (en Europa no hay iv) 30 mg/k/d en 3 dosis durante 1-3 días. Si responde, se aconseja tratamiento de mantenimiento con 30-50 mg/k/d, repartidos en al menos 3 dosis. Hay que vigilar la aparición de posibles efectos secundarios como depresión respiratoria o hipotensión. El pronóstico depende de la instauración precoz del tratamiento.
- Epilepsia dependiente de ácido folínico
Desde 1995 se han descrito varios casos de recién nacidos con crisis intratables que aparentemente respondían a ácido folínico (Hyland et al; 1995). Posteriormente se ha demostrado que estos casos realmente eran epilepsias dependientes de piridoxina (mutaciones en el gen ALDH7A1). Por tanto, el ácido folínico no se considera necesario administrarlo de manera empírica ante un recién nacido con crisis epilépticas (Gallagher et al; 2009). Puede tener un papel coadyuvante en epilepsias dependientes de piridoxina controladas parcialmente.
- Trastornos del metabolismo de la biotina: deficiencia de holocarboxilasa sintetasa y deficiencia de biotinidasa.
Los trastornos del metabolismo de la biotina son un grupo de entidades de transmisión autosómico recesiva que se caracterizan por una deficiencia de las 4 carboxilasas del ser humano: piruvato carboxilasa, propionil-CoA carboxilasa, 3-metilcrotonil-CoA carboxilasa y acetil-CoA carboxilasa.
- Clínica
Hay 2 tipos de defectos del ciclo de la biotina:
1. Deficiencia de carboxilasas múltiples de inicio precoz, o deficiencia de holocarboxilasa sintetasa (HCLS)
La clínica se inicia en las primeras semanas de vida (incluso al nacimiento) en forma de anomalías respiratorias (hiperpnea o apneas episódicas), alteraciones de la conciencia con episodios de letargia que alterna con irritabilidad y alteraciones del tono. Epilepsia en el 25-50%. Pueden presentar otros síntomas generales: rash cutáneo (50%), olor especial de la orina (gato), hipotermia.
2. Déficit de biotinidasa
La clínica suele aparecer en el segundo mes de vida, aunque se han descrito casos de presentación neonatal. Es más frecuente la presencia de crisis mioclónicas refractarias (75%), hipotonía y síntomas cutáneos (hasta el 70% presentan exantema maculo-papular eccematoso y/o alopecia). No es raro que presenten estridor laríngeo o un cuadro de queratoconjuntivitis persistente. Si estos pacientes no son tratados, posteriormente van desarrollando un cuadro de ataxia intermitente, retraso psicomotor, atrofia óptica y sordera neurosensorial. Presentan también mayor susceptibilidad a infecciones.
Ambas entidades suelen presentar episodios agudos intermitentes, en ocasiones graves, de vómitos, letargia y alteración del patrón respiratorio junto con acidosis láctica, cetosis e hiperamoniemia (Wolf; 2010).
- Pruebas complementarias
EEG: brote-supresión o patrón multifocal.
RM cerebral: alteración en la sustancia blanca, dilatación ventricular.
- Diagnóstico
Diagnóstico de sospecha: actividad de biotinidasa en sangre disminuida (generalmente <10%). El 80% presentan aciduria orgánica (con un patrón característico: 3-OH-propiónico, 3 beta-OH-valérico, 3 metilcrotonilglina y metilcítrico).
Diagnóstico definitivo: se realiza por determinación de actividad deficiente de holocarboxilasa sintetasa o biotinidasa en leucocitos o fibroblastos. También se puede realizar por detección de mutaciones en los genes afectos (21q22.1 o 3p25).
- Tratamiento
El tratamiento con biotina (5-20 mg al día) tiene un efecto beneficioso en la resolución de síntomas y anomalías bioquímicas (especialmente en los déficit de biotinidasa) (Cowan et al;2010).
- Deficiencia del cofactor de molibdeno y déficit de sulfito oxidasa
El molibdeno es un oligoelemento esencial para el funcionamiento de tres enzimas: sulfito oxidasa, xantina oxidasa y aldehído oxidasa. Su déficit es ocasionado por una enfermedad autosómica recesiva que puede presentarse de manera conjunta (déficit del cofactor de molibdeno) o de manera aislada en forma de déficit de sulfito oxidasa, con la misma clínica (Palacios et al; 2008).
- Clínica:
Los síntomas consisten en una encefalopatía epiléptica precoz grave aguda o subaguda con leucoencefalopatía cavitante difusa grave (Swaimann; 2012). Desarrollan crisis refractarias mioclónicas en la primera semana de vida, con retraso psicomotor grave, alteraciones del tono, microcefalia adquirida, dificultades para la alimentación y subluxación del cristalino. Suelen presentar un fenotipo peculiar (cara alargada, hipertelorismo, hendiduras palpebrales largas, labios gruesos, filtro amplio). Hay casos con presentación más leve.
- Diagnóstico:
- Diagnóstico de sospecha en deficiencia de sulfito oxidasa (SO): sulfituria (sulfitest positivo) e hipohomocisteinemia.
- Diagnóstico de sospecha de déficit de cofactor de molibdeno (CM): aparte de lo anterior hipouricemia marcada (<1 mg/dl) y aumento de hipoxantina y xantina en orina.
- Diagnóstico de confirmación: determinación enzimática de sulfito oxidasa y xantino oxidasa en fibroblastos. Análisis del gen SUOS (SO) o MOCS, GPHN (CM).
- Pruebas complementarias
- Neuroimagen: leucoencefalomalacia multiquística rápidamente progresiva. El diagnóstico diferencial debe realizarse fundamentalmente con encefalopatía hipóxico isquémica (EHI): pensar en ella en recién nacidos con crisis refractarias e imagen sugestiva de EHI sin antecedentes perinatales compatibles.
- EEG: brote-supresión o trazados multifocales.
- Tratamiento
Ninguno de los tratamientos ensayados ha mostrado una eficacia clara. La mayoría fallecen en los primeros meses de vida.
- Déficit de transportador de glucosa Glut-1
Deficiencia del transportador de glucosa Glut 1; codificado por el gen SLC2A1; necesario para que la glucosa atraviese la barrera hematoencefálica.
- Clínica
La forma clásica (76%) se suele presentar en los primeros 6 meses de vida (raro en periodo neonatal) en forma de encefalopatía epiléptica refractaria (crisis de todo tipo: ausencias, mioclónicas, espasmos, tónicas, atónicas; a veces relacionadas con el ayuno). Posteriormente desarrollan retraso mental, microcefalia, apneas y un trastorno motor complejo con una combinación de espasticidad, distonía y alteración cerebelosa (Keppler et al;2007).
- Diagnóstico
. Diagnóstico de sospecha: la determinación simultánea de glucosa en sangre y LCR sigue siendo la prueba de elección. Para la correcta realización de esta punción lumbar el paciente debe estar en ayuno, y la obtención de sangre se debe realizar inmediatamente antes (15 minutos) que la punción lumbar. Glucosa LCR/Glucosa plasma <0,49 es altamente sugestivo. Hipoglucorraquia (< 40 mg/dL). Lactato en LCR normal o bajo (Wang et al; 2005).
- Diagnóstico de confirmación: análisis molecular del gen SLC2A1.
- Pruebas complementarias
- EEG: paroxismos multifocales, siendo posteriormente frecuentes los complejos punta-onda a 2,5-4 Hz generalizados. Puede mejorar el patrón con la ingesta.
- RM craneal: normal o inespecífica.
- PET con FDG: hipometabolismo difuso en la corteza cerebral y tálamo.
- Tratamiento
Dieta cetogénica, como alternativa energética cerebral, es efectiva en el control de la epilepsia aunque el retraso psicomotor y el nivel cognitivo pueden mejorar sólo parcialmente. Actualmente en experimentación el uso de ácido alfa-lipoico, que aumenta la expresión de GLUT-4. Hay que evitar el uso de fenobarbital y diazepam que disminuyen la función transportadora de GLUT-1.
- Síndrome de Alpers (poliodistrofia infantil progresiva)
Es una encefalopatía rápidamente progresiva que se caracteriza por una epilepsia refractaria generalmente con componente mioclónico y una atrofia cerebral progresiva, con o sin afectación hepática.
Suele iniciarse en periodo del lactante (a partir de los 2 meses); aunque hay casos de presentación neonatal que se manifiestan con un fenotipo característico: síndrome de hipocinesia fetal (retracciones articulares, deformidades torácicas), microcefalia, retrognatia y crecimiento intrauterino retrasado (Frydman et al; 1993).
- Enfermedad de Menkes (tricopoliodistrofia o enfermedad del cabello ensortijado)
Trastorno primario del transporte de cobre, con herencia recesiva ligada a X.
- Clínica
En los 3 primeros meses comienzan con un cuadro de hipotonía, hipotermia y dificultades de alimentación. Son frecuentes las crisis refractarias generalizadas (mioclónicas y espasmos). Se caracterizan por un pelo descolorido, frágil y ensortijado. Pueden presentar malformaciones sistémicas como huesos wormianos, pectus excavatum y diverticulosis intestinal (Turner et al; 2010).
- Exploraciones complementarias:
- Neuroimagen: atrofia cerebral. Arterias cerebrales tortuosas, elongadas y con frecuencia hematomas subdurales.
- EEG: actividad paroxística multifocal.
- Diagnóstico
- Diagnóstico de sospecha: disminución de cobre sérico (<11 micromoles) y la ceruloplasmina (<210 mg/l).
- Diagnóstico de confirmación: secuenciación de de gen ATP7A.
- Tratamiento
La administración de histidinato de cobre parenteral puede prevenir el deterioro neurológico y las crisis, sobre todo si se administra de manera precoz.
- Enfermedades de los neurotransmisores
- Metabolismo del GABA y pterinas
Algunos trastornos de la síntesis de neurotransmisores se manifiestan con un cuadro de crisis refractarias de inicio precoz, fundamentalmente de características mioclónicas. Son fundamentalmente los de síntesis del GABA: déficit de succinil semialdehído deshidrogenasa o aciduria 4-OH butírica (SSDH) y déficit de GABA transaminasa; y los de síntesis de las pterinas: deficiencia de guanosina trifosfatil ciclohidroxilasa(GTPCH) (García-Cazorla et al;2005).
- Déficit de la síntesis de serina
Los defectos en la síntesis de la serina son trastornos autosómicos recesivos que se manifiestan generalmente como un trastorno neurológico precoz y grave: microcefalia congénita, retraso psicomotor, retraso del crecimiento intrauterino, tetraparesia espástica y, a veces, cataratas e hipogonadismo.
El diagnóstico de sospecha se realiza al objetivar niveles de serina disminuidos en LCR (<18 µM/L). Se confirma mediante análisis enzimático en fibroblastos o por análisis mutacional.
Uno de los aspectos más importantes de este trastorno es que el aporte oral de serina puede controlar la epilepsia y mejora el cuadro neurológico (L-serina a 400-600 mg/kg/día en 4 a 6 dosis). Si las crisis persisten se añade glicina 200 mg/kg/d.
- Metabolismo de la homocisteína y los folatos
Los defectos de síntesis de cobalamina o deficiencia de dihidrofolato reductasa se pueden manifestar con crisis de inicio precoz y anemia megaloblástica.
Pero el trastorno que más frecuentemente se presenta con crisis en periodo neonatal de este grupo es el déficit de 5-metilen-tetrahidrofolato reductasa (5MTHFR) que se presenta como un cuadro de encefalopatía epiléptica, microcefalia y apneas. Presenta valores elevados de homocisteína y bajos de metionina. El tratamiento precoz con betaína puede prevenir las secuelas neurológicas (Strauss et al;2007). Además, se puede intentar tratar con riboflavina, hidroxicobalamina y ácido folínico.
- Trastornos de las purinas: déficit de adenilosuccinato liasa (ADSL)
Trastorno autosómico recesivo causado por la deficiencia de la adenilosuccinato liasa que interviene en la síntesis de ATP.
- Clínica
La presentación clínica en periodo neonatal, se caracteriza por un cuadro grave de epilepsia (gran componente mioclónico), retraso del crecimiento intrauterino, microcefalia e hipocinesia fetal. Mortalidad elevada en los primeros meses.
- Pruebas complementarias
RM craneal: en algunos casos atrofia cerebelosa y leucodistrofia.
PET con FDG: disminución de metabolismo glucosa cerebral.
EEG: brote-supresión.
- Diagnóstico
- Diagnóstico de sospecha: test de SAICAR.
- Diagnóstico de confirmación: determinación enzimática y secuenciación del gen ADSL.
- Ceroidolipofuscinosis neonatal
La forma congénita (CLN10) es una entidad autosómica recesiva, causada por la deficiencia de la catepsina D, que es una proteasa lisosomal. Es la forma más grave de ceroidolipofuscinosis. Son recién nacidos con status epilépticos, microcefalia y trastornos respiratorios (apnea). El curso es fatal en días o semanas (Fritchie et al; 2009).
En la neuroimagen se objetiva pérdida de sustancia blanca y gris.
El diagnóstico definitivo se realiza en biopsia de piel en las que se identifican cuerpos de inclusión en las células de Schwann, o en la detección de mutaciones del gen CTSD.
- Defecto del transportador mitocondrial del glutamato
Trastorno autosómico recesivo causado por deficiencia del transportador mitocondrial del glutamato, codificado por el gen SLC25A22. Descrito en 2 pacientes que presentaron epilepsia mioclónica neonatal grave y retinitis pigmentosa. Desarrollan atrofia cerebral. El patrón de EEG muestra brote-supresión. El diagnóstico se realiza mediante análisis del defecto de oxidación del glutamato en fibroblastos y análisis mutacional. No hay tratamiento específico (Molinari et al; 2009).
ECM EN EL QUE LA EPILEPSIA NO ES EL SÍNTOMA PRINCIPAL
- Defectos de betaoxidación de los ácidos grasos
Es frecuente que los trastornos del ciclo de la carnitina (deficiencia de CPT2 y de carnitina-acilcarnitina transferasa), así como la aciduria glutárica tipo 2 presenten crisis.
- Trastornos peroxisomales
Son frecuentes las crisis en el síndrome de Zellweger (hasta en el 92%), adrenoleucodistrofia neonatal, la condrodisplasia punctata rizomélica y en algunos defectos monoenzimáticos (deficiencia de acil-CoA oxidasa, deficiencia de proteína bifuncional). Las crisis son de todo tipo y el patrón EEG suele ser multifocal.
- Aminoacidopatías, trastornos del ciclo de la urea y acidurias orgánicas
Son cuadros de encefalopatía aguda que pueden presentar crisis. Destacar que algunas aminoacidurias y acidemias congénitas pueden causar posturas distónicas o movimientos atetoides repetitivos (eventos como “pedaleo” o “boxeo” se describen con frecuencia) que pueden simular episodios convulsivos y que a veces cuesta diferenciar de las crisis epilépticas. Dentro de estos trastornos, los que más frecuentemente asocian crisis son:
- Aminoacidopatías: hiperglicinemia no cetósica, leucinosis, fenilcetonuria (25%).
- Acidemias orgánicas: acidemia isovalérica, acidemia propiónica, acidemia metilmalónica, aciduria glutárica tipo 1, deficiencia de 2-metil-3-hidroxibutiril-CoA deshidrogenasa y deficiencia de 3-metilcrotonil-CoA carboxilasa. A destacar la aciduria D2 hidroxiglutárica que se manifiesta como una encefalopatia epiléptica neonatal refractaria (Struys et al 2005).
- Crisis secundarias a alteraciones electrolíticas
En un neonato con crisis siempre se debe descartar hipoglucemia, pues se puede tratar con facilidad con glucosa intravenosa. En el recién nacido se debe descartar sepsis, enfermedad sistémica grave y diabetes materna. Las crisis prolongadas secundarias a hipoglucemia pueden producir lesión del lóbulo occipital. La hipoglucemia en el prematuro normalmente se debe a problemas de adaptación y no suele requerir amplios estudios. En ocasiones puede ser necesario descartar glucogenosis hepática, defectos de gluconeogénesis, defectos de betaoxidación, hiperinsulinismo congénito, aminoacidopatías y acidurias orgánicas. Para ello en el momento de la hipoglucemia se deben determinar: glucosa, cetonemia, ácidos grasos libres, láctico, amonio, aminoácidos, acilcarnitinas, insulina, cortisol y GH en plasma; así como cetonuria y ácidos orgánicos en orina.
Otras causas de crisis corregibles son las secundarias a hipocalcemia, hipomagnesemia o hipernatremia.
- Trastornos mitocondriales
Se asocian con frecuencia a epilepsia en 25-60%, especialmente en el síndrome de Leigh (50%). En general, las crisis son más frecuentes en aquellos con enfermedad de inicio precoz y retraso psicomotor grave, y menos frecuentes en aquellos con enfermedad más leve. Clínicamente se ven todo tipo de crisis, pero especialmente frecuentes son las que tienen componente mioclónico o espasmos (Sadleir et al;2004).
- Trastornos lisosomales
Son pocas las enfermedades lisosomales que presentan crisis en el periodo neonatal: enfermedad de Krabbe, deficiencia de GM3 sintasa y deficiencia de prosaposin (Staretz-Chacham et al;2009).
BIBLIOGRAFÍA
- Baxter P. (2001). Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol. 43:416-20.
- Cowan T, Blitzer MG, Wolf B. (2010). Working Group of the American College of Medical Genetics Laboratory Quaity Assurance Committee, Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med. 12:464-470.
- Fritchie K, Siintola E, Armao D, Lehesjoki AE, Marino T et al. (2009). Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol. 117:201-208.
- Frydman M, Jager-Roman E, de Vries L, Stoltenburg-Didinger G, Nussinovitch M et al. (1993). Alpers progressive infantile neuronal poliodystrophy: an acute neonatal form with findings of the fetal akinesia syndrome. Am J Med Genet. 47:31-36.
- Gallagher RC, Van Hove JLK, Scharer G, Hyland K, Plecko B et al. (2009). Folinic acid-responsive seizures are identical to piridoxine-dependent epilepsy. Ann Neurol. 65:188-194.
- García-Cazorla A, Ormazábal A, Artuch R, Pérez-Dueñas B, López-Casas J et al. (2005). Errores congénitos de los neurotransmisores en neuropediatría. Rev Neurol. 41:99-108.
- Gospe SM Jr. (1998). Current perspectives on pyridoxine-dependent seizures. J Pediatr. 132:919-23.
- Hoffmann GF, Schmitt B, Windfuhr M, Wagner N, Strehl H et al. (2007). Pyridoxal 5´phosphate may be curative in early-onset epileptic encephalopathy. J Inherited Metab Dis. 30:96-99.
- Hyland K, Buist NRM, Powell BR, et al. (1995). Folinic acid responsive seizures: a new syndrome?. J Inherit Metab Dis.18:177-81.
- Jurecka A, Zikanova M, Tylki-Szymanska A, Krijt J, Bogdanska W et al. (2008). Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol Genet Metab. 94:435-442.
- Klepper J, Leiendecker B. GLUTl deficiency syndrome. (2007). Dev Med Child Neurol. 49:707-16.
- Martínez-Bermejo A, LópezMartín V, Arcas J, Tendero A, Roche C et al. (2001). Coma alfa: correlación clínica, electroencefalográfica y etiológica en la edad pediátrica. Rev Neurol. 33:1101-1105.
- Molinari F, Kaminska A, Fiermonte G, Boddaert N, Raas-Rothschild A. (2009). Mutations in the mitocondrial glutamate carrier SLC25A22 in neonatal epileptic encephalolophaty with suppressions bursts. Clin Genet. 76:188-194.
- Parikh S, Hyland K, Lachhwani DK. (2009). Vitamins, not surgery: Spinal fluid testing in hemispheric epilepsy. Pediatr Neurol.40:477-479.
- Palacios A, García MT, Sanchez J, Nogales A, Puche A. (2008). Déficit del cofactor molibdeno como causa de encefalopatía epiléptica precoz. An Pediatr (Barc). 69:181-196.
- Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E et al. (2007). Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat. 28:19-26.
- Sadleir LG, Connolly MB, Applegarth D, Hendson G, Clarke L et al. (2004). Spasms in children with definite and probable mitochondrial disease. 11:103-110.
- Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. (2009). Lysosomal storage disorders in the newborn. Pediatrics. 123:1191
- Strauss KA, Morton DH, Puffneberger EG, Hendrickson C, Robinson DI, Wagner C et al. (2007). Prevention of brain disease from severe 5,10-metylenetetetrahydrofolate reductase deficiency. Mol Genet Metab 91:165-167.
- Struys EA, Salomons GS, Achouri Y, Van Schaftingen E, Grosso S, Craigen WJ et al. (2005). Mutations in the D2-hydroxyglutarate dehydrogenase gene cause D2-hydroxiglutaric aciduria. Am J Hum Genet. 76:358-360.
- Turner Z, Moller LB. (2010). Menkes disease. Eur J Hum Genet. 18:511-518.
- Van Hove J, Lohr NJ. (2011). Metabolic and monogenic causes of seizures in neonates and young infants. Mol Genet Metab. 104:214-220.
- Wang D, Pascual JM, Yang H, et al. (2005). Glut-1 deficiency syndrome: clinical, genetic and therapeutic aspects. Ann Neurol. 57:111-8.
- Wolf B. (2010). Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 100:6-13.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}