Trastornos del metabolismo del GABA
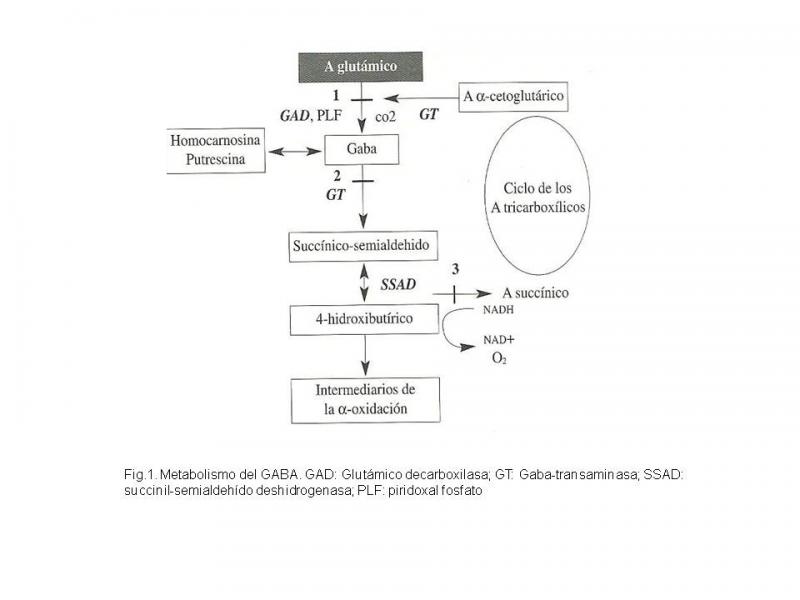
El GABA es el NT más abundante con una función preferentemente inhibitoria. La vía metabólica del GABA aparece representada en la Fig.1.
Fig.1. Metabolismo del GABA. GAD: Glutámico decarboxilasa; GT: Gaba-transaminasa; SSAD: succinil-semialdehído deshidrogenasa; PLF: piridoxal fosfato
Dentro de este grupo merecen especial interés el déficit de la succínil semialdehido-deshidrogenasa (SSAD) y de la transaminasa del ácido ɣ-aminobutírico (GT).
- DÉFICIT DE SUCCÍNIL SEMIALDEHIDO-DESHIDROGENASA (SSAD)
Enfermedad de herencia autosómica recesiva, también conocida como aciduria ɣ-hidroxibutírica.
Clínica: retraso en el desarrollo, hipotonía, hiporreflexia, retraso en el lenguaje expresivo, retraso mental, ataxia no progresiva, trastornos de conducta (hiperactividad, ansiedad, déficit atencional, agresividad…), trastornos de sueño y convulsiones. Puede observarse atrofia cerebral y/o cerebelosa e hiperintensidad en T2 en ganglios basales en RMC.
Diagnóstico bioquímico: elevación de ɣ-hidroxibutírico en LCR y plasma, sin acidosis metabólica, hipoglucemia ni hiperamoniemia. Se puede realizar diagnóstico enzimático en linfocitos o linfoblastos.
Tratamiento: se ha empleado la Vigabatrina como inhibidor irreversible de la enzima GT o la Lamotrigina como inhibidor de la acción del glutamato, con resultados clínicos variables. Debe evitarse el uso de Valproato. Los ISRS o el metilfenidato pueden ser útiles en los trastornos de conducta.
- DEFICIT DE TRANSAMINASA DEL ÁCIDO Ɣ-AMINOBUTÍRICO (GT)
Enfermedad de presentación rara y herencia autosómica recesiva.
Clínica: encefalopatía epiléptica progresiva, retraso en el desarrollo, hipotonía y macrosomía.
Diagnóstico: En la RMC se pueden detectar trastornos de la migración neuronal, agenesia de cuerpo calloso o hipoplasia cerebelosa y en la RM espectroscópica se puede detectar elevación de GABA cerebral. El diagnóstico de laboratorio viene determinado fundamentalmente por la elevación de GABA libre en LCR y plasma.
Tratamiento: no existe.
BIBLIOGRAFIA
- Echenne B, Roubertie A, Assmann B, Lutz T, Penzien JM, Thony B et al. (2006). Sepiapterin reductase deficiency: clinical presentation and evaluation of long term therapy. Pediatr Neurol. 35: 308-13.
- Forsyth R, Newton R. (2007). Paediatric Neurology. New York: Oxford University Press.
- Friedman J, Hyland K, Blau N, MacColin M. (2006). Dopa-responsive hypersomnia and mixed movement disorders due to sepiapterin reductase deficiency. Neurology. 67: 2232-36.
- García-Cazorla A, Ormazábal A, Artuch R, Pérez-Dueñas B, López-Casas J, Fernández-Álvarez E, Campistol J. (2005). Errores congénitos de los neurotransmisores en Neuropediatría. Rev Neurol. 41: 99-107.
- Hoffmann GF, Zschocke J, Nyhan WL. (2010).Inherited Metabolic Diseases. A Clinical Approach. Berlin: Springer.
- Hyland K. (2003). Disorders of neurotransmitter metabolism. En: Blau N, Duran M, Blaskovics ME, Gibson KM, Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases, (pp 107-22).
- Jaeken J, Jakobs C, Clayton PT, Wevers RA. (2006).Disorders of Neurotransmission. En: Fernandes J, Saudubray JM, Van den Berghe G, Walter JH. Inborn Metabolic Diseases.Diagnosis and Treatment, (pp 357-72). Berlin: Springer-Verlag.
- Lyon G, Adam RD, Kolodny EH. (1996). Neurology of hereditary metabolic diseases of children. New York: McGraw-Hill.
- Neville BG, Parascandalo R, Farrugia R, Felice A. (2005). Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain. 128: 2291-6.
- Sanjurjo P, Baldellou A. (2010). Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon.
- Verdú A, Cazorla MR. (2008). Trastornos del metabolismo intermediario con repercusión neurológica (II). En: Verdú A, García A, Martínez B. Manual de neurología infantil, (pp 622-34). Madrid: Publimed.
- Zschocke J, Hoffman GF. (2004). Vademecum metabolicum: manual of metabolic paediatrics. Friedrichsdorf: Schattaauer.

{kind=link}