Metabolopatía neonatal con hipotonía
INTRODUCCIÓN
La hipotonía es un signo muy inespecífico en el recién nacido enfermo, siendo casi constante en enfermedades no metabólicas (encefalopatías hipóxico-isquémicas, enfermedades musculares…). Casi el 60% de los recién nacidos con un error congénito del metabolismo (ECM) presentan hipotonía central, aunque suele ser un síntoma más dentro de un contexto generalizado de afectación neurológica. Sólo unos ECM pueden presentarse como hipotonía aislada en periodo neonatal:
1.- Fundamentalmente los ECM del tipo alteración del déficit energético:
- Defectos de beta oxidación.
- Enfermedades mitocondriales.
2.-También los ECM de alteración de las moléculas complejas:
- Peroxisomales.
- Síndrome de glicoproteínas deficientes en carbohidratos (CDG).
- Errores innatos de la síntesis del colesterol.
3.- Enfermedad de Pompe.
4.- Otros trastornos: trastornos del ciclo de la urea, déficit de sulfito oxidasa, Menkes, hiperglicinemia no cetósica.
PRINCIPALES ECM QUE SE MANIFIESTAN COMO HIPOTONÍA EN EL PERIODO NEONATAL
1.- Síndrome de glicoproteínas deficientes en carbohidratos (CDG).
Es un grupo heterogéneo de enfermedades causadas por defectos en la síntesis de glucanos, en su unión a otros compuestos (proteínas y lípidos) y/o en el procesamiento posterior de los glucoconjugados; procesos que se producen en el retículo endoplásmico y aparato de Golgi. Se han descrito hasta el momento al menos 25 tipos de CDG que se corresponden cada uno de ellos a defectos enzimáticos diferentes (Vilaseca et al; 2004) (Van Hove et al; 2004).
1.1.- Clínica
Gran heterogeneidad; ha de sospecharse en pacientes con trastornos multisistémicos. Cuando se presentan en periodo neonatal es frecuente un cuadro neurológico de hipotonía, alteración de la conciencia, crisis y dismorfia (lipodistrofias y mamilas invertidas). Pueden presentar trastornos de la coagulación (sangrado o “stroke-like”), y otros síntomas sistémicos como hepatopatía, enteropatía, ictiosis…
El tipo más frecuente es el Ia debido a una deficiencia de la enzima fosfomanomutasa (PMM); que se presenta en periodo neonatal fundamentalmente con hipotonía, dismorfia, cardiomiopatía con efusión pericárdica y transaminasas elevadas. Se produce por mutaciones en el gen PMM2. Es frecuente el hallazgo de atrofia cerebelosa precoz en la neuroimagen (Jaeken et al; 2003).
-
- 1.2.- Diagnóstico
- Diagnóstico de sospecha: disminución de glicoproteínas (ceruloplasmina, haptoglobina, factores de coagulación, etc). Aumento de la transferrina deficiente en carbohidratos (CDT). Electroforesis de transferrina (CDG de tipo I o II).
- Diagnóstico de confirmación: estudio enzimático en fibroblastos o secuenciación génica.
1,3.- Tratamiento
Sintomático
2.- Enfermedades peroxisomales
Los peroxisomas son organelas que se encuentran en todas las células nucleadas con múltiples funciones: síntesis de plasmalógenos, beta-oxidación de ácidos grasos de cadena muy larga (AGCML), oxidación de ácido fitánico, formación de ácidos biliares y reacciones dependientes de catalasa. Las enfermedades peroxisomales son alteraciones autosómicas recesivas debidas a la disfunción de los peroxisomas. Existe una gran variabilidad clínica y se pueden presentar en todas las edades, vamos a describir las que se inician en el periodo neonatal (Lazarow et al; 1995):
2.1.- Clínica
Se clasifican en dos grupos (Lyon et al; 2006):
2.1.1.- Trastornos de la biogénesis peroxisomal
Ausencia virtual de peroxisomas y disfunción de las múltiples enzimas que contienen los peroxisomas.
2.1.1.1.- Trastornos del espectro Zellweger: Forman un espectro continuo de la misma enfermedad que se diferencian por la severidad y la presencia de determinadas características clínicas. Se producen por alteración de los genes PEX.
2.1.1.1.1.- Síndrome de Zellweger (SZ)
Encefalopatía crónica de inicio en el periodo neonatal que se caracteriza fundamentalmente por hipotonía grave (con hiporreflexia), dismorfia craneofacial (fontanela grande, frente alta, arcos supraorbitarios hipoplásicos, puente nasal bajo y ancho, epicantus, oblicuidad mongoloide y orejas anormales), ausencia de desarrollo psicomotor, crisis epilépticas de todo tipo (80%), anomalías oculares (retinitis pigmentaria, opacidad corneal) y sordera. También son frecuentes la hepatomegalia, insuficiencia adrenal, colestasis, quistes renales, calcificación patelar y anomalías de la migración neuronal. Fallecen en los primeros meses (media: 7 meses).
2.1.1.1.2.- Adrenoleucodistrofia neonatal (ALD)
Forma intermedia. La enfermedad es menos grave y suelen alcanzar la adolescencia precoz, algunos consiguen marcha autónoma (generalmente con ataxia), y decir algunas palabras, pero tienen un grave retraso (mortalidad media 36 meses). Se caracteriza por hipotonía, crisis, sordera y degeneración retiniana. La dismorfia facial es menos marcada o ausente.
2.1.1.1.3.- Refsum infantil.
La forma más leve del espectro. Tras un periodo neonatal normal, a los 1-3 años desarrollan retraso mental moderado-grave con hipotonía, ataxia, retinopatía y sordera neurosensorial. El curso es mucho más prolongado: la mayoría llegan a la niñez tardía, adolescencia o vida adulta. No presentan crisis.
2.1.1.2.- Condrodisplasia punctata rizomélica (CDPR)
Se caracteriza por 3 defectos bioquímicos: síntesis de plasmalógenos, oxidación de ácido fitánico y tiolasa peroxisomal. Está presente la reacción catalasa positiva. Se produce por mutación en el gen PEX7.
Se caracterizan por: acortamiento de los segmentos proximales de los miembros, contracturas articulares, cataratas congénitas, dismorfia facial, crisis y desarrollo de retraso psicomotor grave. Mortalidad en la infancia temprana. Algunos pueden presentar ictiosis al nacimiento.
2.1.2.- Defectos monoenzimáticos
Peroxisomas de aspecto normal, con defecto enzimático único de la β-oxidación peroxisomal. Menos frecuentes. Suelen presentar hipotonía, crisis y dismorfia facial tipo Zellweger. Incluyen: deficiencia de 3-oxoacil-CoA tiolasa (o pseudoZellweger), deficiencia de acil-CoA oxidasa (o pseudoALD neonatal), deficiencia de la enzima bifuncional (DPB), deficiencia de DHAP-AT (o RCDP2) y deficiencia de alkil-DHAP sintasa (o RCDP3).
2.2.-Pruebas complementarias
- Neuroimagen: Todos menos la CDPR presentan afectación de la sustancia blanca.
- Ecografía abdominal: son frecuentes los quistes renales en el SZ, pseudo-SZ y DPB.
- Radiología simple: puede mostrar anomalías óseas como calcificaciones patelares y sincondrosis del acetábulo en SZ; o calcificaciones puntiformes epifisarias y extraepifisarias, más diseminadas en CDPR.
- EEG: anomalías multifocales.
2.3.- Diagnóstico
Diagnóstico de sospecha: todos los trastornos peroxisomales, salvo la CDPR, presentan un aumento de los ácidos grasos de cadena muy larga (AGCML). Para poder diferenciarlos es necesario determinar los plasmalógenos eritrocitarios, ácido fitánico, ácido pristánico, ácido pipecólico y metabolitos intermediarios de la síntesis de los ácidos biliares.
Diagnóstico de confirmación: es posible estudiar con microscopía electrónica los peroxisomas hepáticos. Determinación genética (PEX, PEX7). Para el diagnóstico de las peroxisomopatías monoenzimáticas es necesario la determinación enzimática correspondiente en fibroblastos y/o hígado
2.4.- Tratamiento
En pacientes con trastornos peroxisomales generalizados es fundamentalmente sintomático: vitamina K, terapia hormonal sustitutiva, ácido ursodeoxicólico (puede disminuir los niveles de intermediarios de los ácidos biliares) y suplementos de vitaminas liposolubles. Los intentos de normalizar algunas anomalías bioquímicas mediante éter lípido oral, restricción dietética de AGCML y fitánico no han demostrado eficacia clínica.
El empleo dietético de ácido docosahexanoico (DHA) en SZ restaura los niveles deficientes en plasma y hematíes, al parecer con mejoría clínica parcial en los casos menos graves.
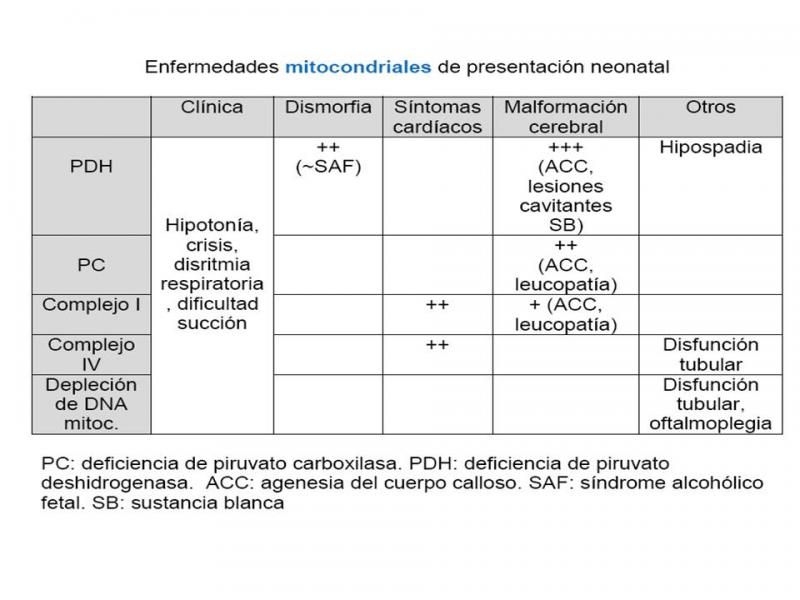
3.- Enfermedades mitocondriales
Los trastornos mitocondriales se manifiestan por un trastorno multisistémico que puede afectar a diversos órganos: corazón, músculo, cerebro y sistema endocrino. Cuando se presenta en periodo neonatal, son cuadros clínicos graves con evolución fatal en meses, con síntomas de hipotonía grave, patrones respiratorios anormales (apneas), crisis (25-60%) y letargia. Pueden asociar afectación cardíaca, hepática, dismorfia facial y quistes renales.
Las enfermedades que se pueden presentar en periodo neonatal son fundamentalmente:
- Trastornos del metabolito del piruvato: deficiencia de la piruvato deshidrogenasa (PDH) o piruvato carboxilasa (PC)
- Trastornos de los complejos de la cadena respiratoria mitocondrial I (NADH-ubiquinona oxidoreductasa) y IV (citocromo C-oxidasa)
- Síndrome de depleción del DNA mitocondrial.
- Diagnóstico
- Diagnóstico de sospecha: aumento de ácido láctico en sangre o LCR. Cetonemia (en déficit de PC, no en PDH ni CRM). El índice lactato/piruvato se encuentra elevado (>20) excepto en PDH. Pueden presentar hipoglucemia e hipertransaminasemia.
- Diagnóstico de confirmación: actividad enzimática en fibroblastos (y músculo en CRM). Secuenciación genética cuando se conozca el gen afecto.
- Síndrome de Leigh
Una forma de presentación frecuente es el síndrome de Leigh (encefalopatía necrotizante subaguda) que se caracteriza por afectación simétrica de los ganglios basales, tronco, y núcleo dentado del cerebelo. Clínica similar a los previos, aunque son más frecuentes las crisis (50%). Puede deberse a varios defectos metabólicos: trastorno del complejo I, II y IV, V (gen ATP6), síndromes de depleción mitocondrial (SUCLA2, POLG) y déficit de PDH.
- Tratamiento
Sintomático en la mayoría de las ocasiones. En PDH: dieta cetógenica y en ocasiones puede ser útil la tiamina. En PC: se ha intentado tratamiento en ocasiones efectivo con triheptanoino, aspartato o citrato.
BIBLIOGRAFÍA
- Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O et al. (2010). Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol 52:916-921.
- Jaeken J. (2003). Congenital disorders of glycosilation (CDG): It´s all in it! J Inherit Metab Dis. 26:99-118.
- Lazarow PB, Moser HW. (1995). Disorders of peroxisome biogenesis. En: Scriver CR, Beaudet Al, Sly WS et al (ed). The Metabolic and Molecular Bases of Inherited Disease. (pp 2287-2324). New York: McGraw-hill
- Lyon G. (2006). The neurology of neonatal hereditary metabolic diseases. En: Lyon G, Kolodny EH, Pastores GM (Ed), Neurology of hereditary metabolic diseases of children, (pp9-64). McGraw-Hill.
- Van Hove J, Lohr NJ. (2011). Metabolic and monogenic causes of seizures in neonates and young infants. Mol Genet Metab 104:214-220
- Vilaseca M, Artuch R, Briones P. (2004). Defectos congénitos de la glucosilación: últimos avances y experiencia española. Med Clin (Barc). 122:707-16.

{kind=link}
{kind=link}